New deep learning technology using H&E images has created an alternative path for molecular diagnostics
Breast cancer is a clear example of the effectiveness of precision medicine. Tumors that test positive for the HER2 protein can be targeted with immunotherapy, radically improving the prognosis. Treatments for many cancer types may be effective only for specific mutations or genomic profiles. Understanding the subset of patients who may benefit is the key to personalized cancer treatments.
However, the methods to assess molecular properties of tumors are expensive and time-consuming. Each new test also requires additional tissue. Recent advances in computational pathology have found an alternative: machine learning algorithms applied to H&E histology. Dozens of studies- all published in the last few years - have demonstrated that one or more molecular properties can be predicted from H&E alone using the advancements of deep learning.
The Limitations of Molecular Testing
Molecular and genomic properties can be used to stratify patients into subtypes. Each subtype responds to some courses of treatment but not others and tends to have a distinct prognosis. Advances in precision medicine over the last decade have focused on these molecular biomarkers, which have helped identify the significant proportion of patients who don’t respond to immunotherapy.
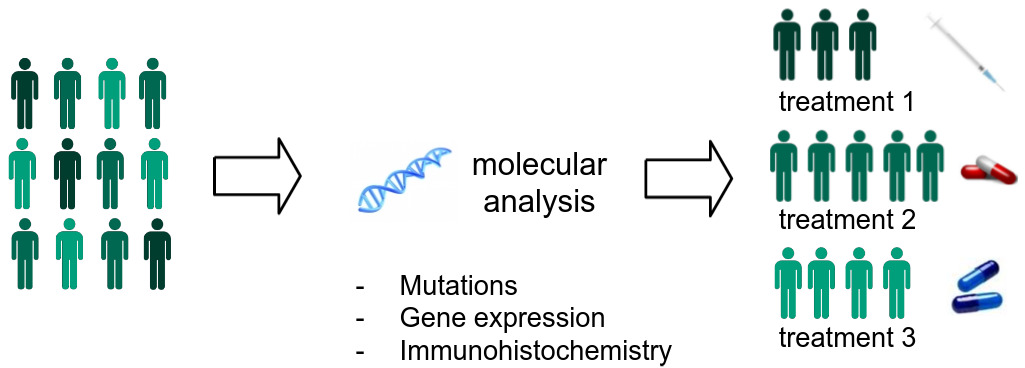
The technologies used in assessing these molecular properties are expensive and time-consuming to perform. They may involve DNA sequencing to detect mutations, RNA sequencing to assess gene expression, or immunohistochemical staining to identify protein biomarkers. Most are not routinely performed on all patients who could benefit and are not done at all in labs with limited resources. To complicate things further, in many cases only a small amount of tissue is excised from a tumor, and there is not enough for additional analyses beyond what a pathologist examines through the microscope.
New studies keep identifying more molecular properties of potential clinical value, each requiring its own tissue sample and processing procedure. Current workflows are not designed to incorporate this many tests. While comprehensive molecular testing will be difficult to implement at scale, histological staining of tissue is common practice and imaging of such samples is becoming increasingly available with the transition to digital pathology.
Deciphering Histological Signatures with Deep Learning
Digital pathology alone is not sufficient to assess molecular biomarkers as their histological signatures are often too complex to be recognizable by pathologists. But recent advances in deep learning can now decipher patterns that are beyond the limits of human perception.
Four types of molecular biomarkers have been successfully predicted from H&E in studies of more than ten different types of cancer:
- protein biomarkers
- genomic subtypes and expression of individual genes
- molecular alterations (mutations, microsatellite instability, chromosomal instability, tumor mutational burden, DNA repair deficiency)
- virus
This article will review the multitude of models that have been developed to predict molecular biomarkers. From bottom-up models based solely on deep learning to somewhat more interpretable or pathologist-driven models that capture aspects of tissue morphology or the tumor microenvironment.
Deep Learning Methods for Whole Slides
Predicting molecular tumor properties from H&E is an exploding area of research.
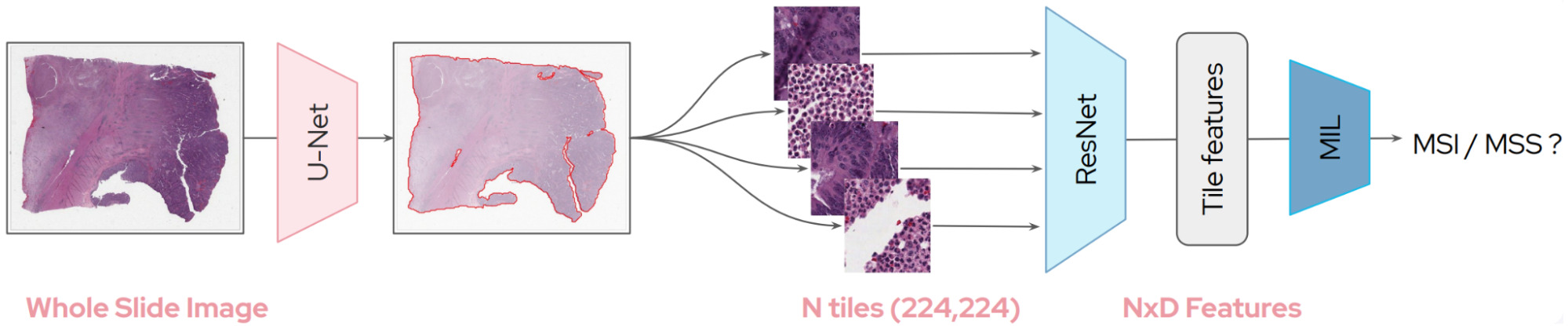
Whole slide images (WSIs) are massive and time-consuming to annotate in detail. For this reason, most algorithms to predict molecular biomarkers rely on weak supervision in which only a patient-level label is available for training.
This weak supervision makes it challenging to learn a representation for image patches and so most models have relied upon transfer learning with a model pretrained on the ubiquitous dataset of computer vision: ImageNet.
Tile Selection
The first decision in processing WSIs is selecting which tiles to use in a model. WSIs can be 100,000x100,000 pixels or larger. Tiles containing mostly background can easily be discarded. But what about the rest?
Some studies have relied upon manual annotations of tumor regions from pathologists. This could take the form of tissue microarray cores that are selected by pathologists [Couture2018a, Couture2018b, Rawat2020, Shamai2019] or annotated WSIs [Kim2020, Popovici2017].
Others have elected to develop a tumor vs. non-tumor model, either to classify each tile or to compute a pixel-level segmentation for the WSI [Kather2019a, Xu2020].
Depending on the model, all tumor tiles may be used to train a biomarker model or some subset.
Some studies select a random subset of tiles [Hohne2021], others are more strategic. For example, Xu et al. selected representative tiles by clustering all tiles and picking tiles from each cluster [Xu2020].

While most studies train and test models only on tumor tissue, others have experimented with including non-tumor tiles.
Rawat et al. compared models that used all tissue tiles, epithelium only, stroma only, fat only, or epithelium and stroma for predicting breast cancer receptor status [Rawat2020]. The best results were achieved using epithelium and stroma but models using all patches or epithelium only were only 1% behind.
Tile Aggregation
So at this point we have a selection of tiles from each WSI. Typically, each is assigned the patient-level label, for example, positive or negative for a particular biomarker. Then a Convolutional Neural Network (CNN) is trained using the set of patches and their corresponding label.
But how do we then make patient-level predictions at inference time?
The most common strategy is a simple aggregation of tile predictions using the mean prediction or taking a majority vote [Coudray2018,Kather2019b, Fu2020].

This approach will work well in some cases, but what if the tumor is heterogeneous or not all tiles are informative of the biomarker status?
Somewhat more robust strategies involve a second stage classifier to make patient-level predictions.
La Barbera et al. calculated statistics on the tile predictions to form a single patient-level feature vector, on which the second stage classifier was trained to predict HER2 status of breast cancer [LaBarbera2020].
Alternatively, a quantile function can represent the distribution of tile predictions [Couture2018a]. A second classifier is then trained using the quantile function for each tumor and used to predict the final class. Quantile functions can also be incorporated into a CNN as a pooling layer so that the whole model may be trained end-to-end [Couture2018b].
Attention-based Aggregation
Rather than training a tile classiifer and a second stage model to aggregate tile predictions, most research now favors a model to aggregate tile features using attention [Hohne2021, Abbasi-Sureshjani2021, Schirris2021, Saillard2021, Anand2021]. Following DeepMIL by Ilse et al., tile features are combined as a weighted sum, where the attention weights are learned by the model itself from the tile features [Ilse2018]. This way the model can place more weight on tiles that are informative for the specific task.

An attention model can be applied as a CNN aggregation layer for end-to-end prediction. In order to fit the entire model on a GPU for training, a subset of patches must be selected from each WSI. For example, Hohne et al. selected 30 tiles from each WSI of thyroid cancer, with this selection made randomly for each epoch while training to predict mutation status [Hohne2021].
Alternatively, each tile may be encoded by a pretrained model, compressing it down to a smaller representation [Abbasi-Sureshjani2021, Schirris2021, Saillard2021]. This way, the encoded features for the entire slide may fit on the GPU, and the attention model is learned using all tiles.
Self-Supervised Learning of Tile Features
The simplest means to encode a tile is with a CNN pretrained on ImageNet. While the features are not tuned to histology images, they are still quite powerful – particularly the lower CNN layers.
Recent advances in self-supervised learning now present an alternative: pretrain a model on patches from WSIs with a pretext task in order to learn a better representation for histology.
A detailed review of the pretext tasks used for pathology goes beyond the scope of this article (a likely topic for a future article though!). But the most commonly used pretext tasks take a contrastive or a clustering objective.
Rawat et al. created an image representation called “fingerprinting” that uses self-supervision to learn features that distinguish one patient from another [Rawat2020]. They demonstrated that this representation works better than a fully supervised approach for predicting breast cancer receptor status.
Weakly Supervised Learning with Self-Supervised Features
Abbasi-Sureshjani et al. used attention-based deep multiple instance learning with a backbone CNN pretrained in one of two ways: on ImageNet or on histology images with the contrastive learning method BYOL [Abbasi-Sureshjani2021].
In most cases, the two backbones performed similarly for breast cancer molecular subtypes. However, the self-supervised one was significantly better when the model was tested on slides from a different scanner than the model was trained on.
Saillard et al. compared three different frameworks for predicting microsatellite instability using either an ImageNet pretrained CNN or a contrastive one using the method MoCo [Saillard2021]:
- A simple average pooling of features across tiles.
- Chowder, which applies a neural net to each tile and then takes the tiles with the top N and bottom N scores for the final model.
- DeepMIL, an attention-based deep multiple instance model.
The best multiple instance framework varied somewhat with the dataset; however, the self-supervised variants produced better results than the ImageNet ones, especially when applying the model to a different cohort.
The self-supervised models also provided a clearer interpretation for pathologists of the patterns learned by the model.

The improved generalization performance found by Abbasi-Sureshjani et al. [Abbasi-Sureshjani2021] and Sailard et al. [Saillard2021] demonstrates an interesting potential for self-supervised learning.
Incorporating Tumor Heterogeneity
While most tumors likely belong to a single class, others may be heterogeneous. Some portions may have a particular mutation while the rest does not, or more than one molecular subtype can be present.
Schirris et al. took the DeepMIL framework a step further to incorporate a measure of heterogeneity [Schirris2021].
They pretrained a CNN using the self-supervised learning method SimCLR, then used it to encode tiles from WSIs.
The tile representations are combined with a deep multiple instance learning model called DeepSMILE (Self-supervised heterogeneity-aware Multiple Instance LEarning). This model incorporates an attention module like DeepMIL and also integrates a measure of feature variance to capture heterogeneity.
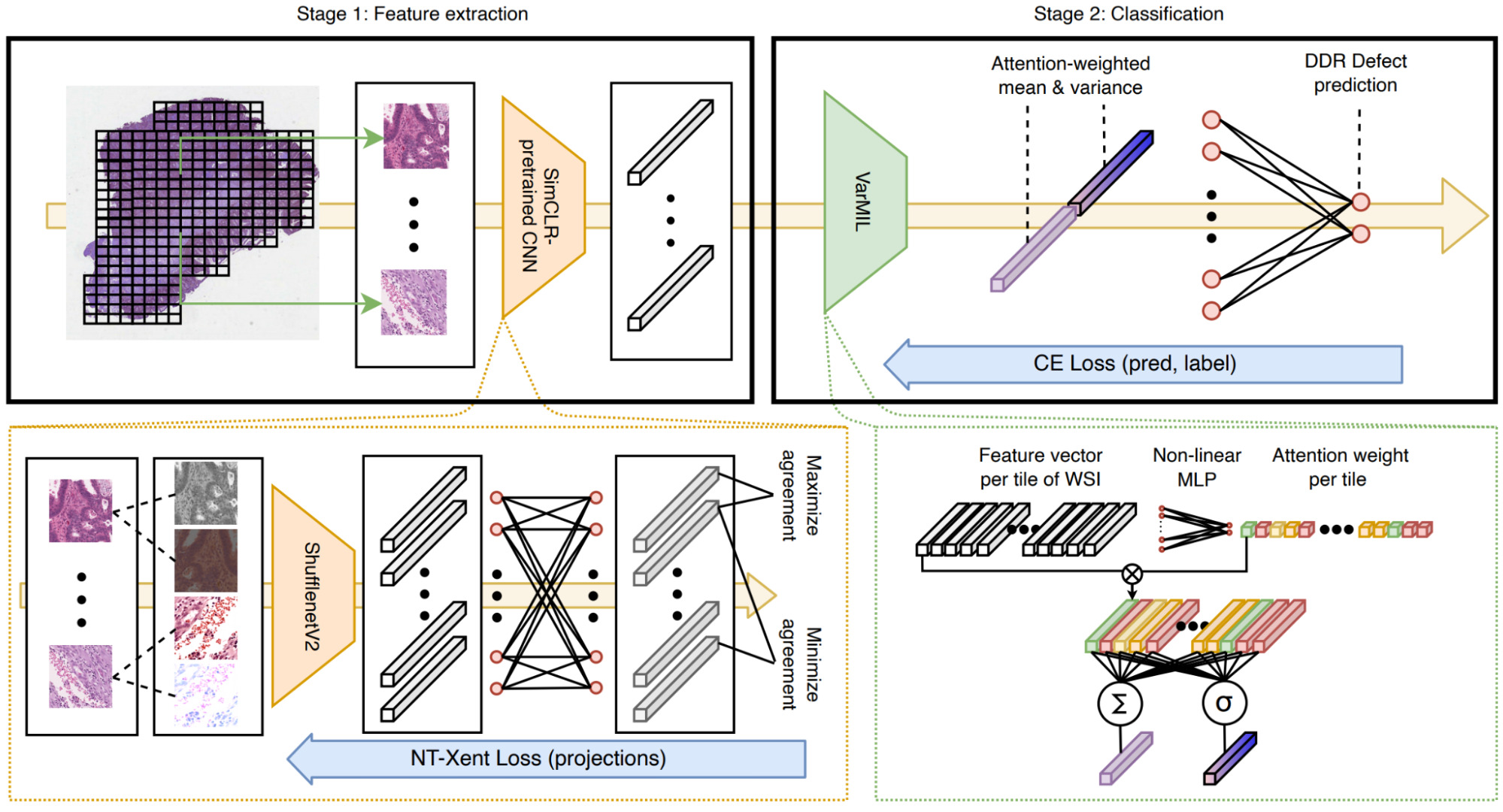
Once again, self-supervised pretraining produced a significant improvement in model performance compared to a model pretrained on ImageNet. The attention-based multiple instance component also improved performance significantly with an additional small boost from attention-weighted variance.
Pretraining with Gene Expression
Self-supervised learning is one powerful method for pretraining a CNN. Schmauch et al. proposed another: pretraining by predicting RNA-Seq gene expression [Schmauch2020].
They first trained their HE2RNA model to predict gene expression. Predictions for some genes were more accurate than others, and this model alone produced some fascinating results.
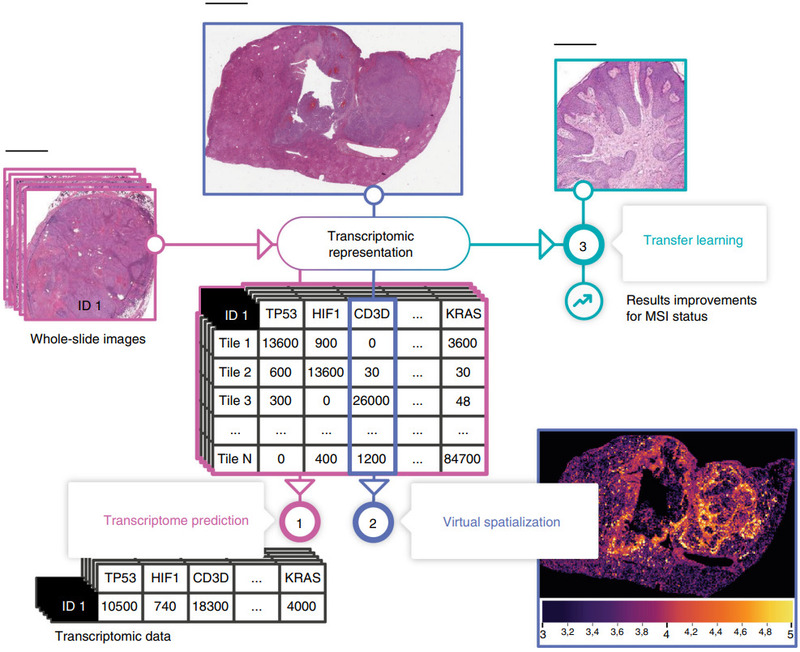
But then they transferred the gene expression model to a much smaller data set to predict microsatellite instability.
Gathering a sufficiently large histology data set for training a deep learning model is challenging. HE2RNA could provide a means to pretrain on a large data set like TCGA and finetune on a data set with fewer patients.
Other studies have also predicted gene expression directly. Levy-Jurgenson et al. trained models to predict mRNA and miRNA expression for breast and lung cancer [Levy-Jurgenson2020], while Wang et al. studied mRNA expression for more than 17 thousand genes in breast cancer [Wang2021b].
Validating Weakly Supervised Models
Weakly supervised models can be validated at the patient- or slide-level, but it is much more difficult to validate the heatmaps they produce with localized predictions.
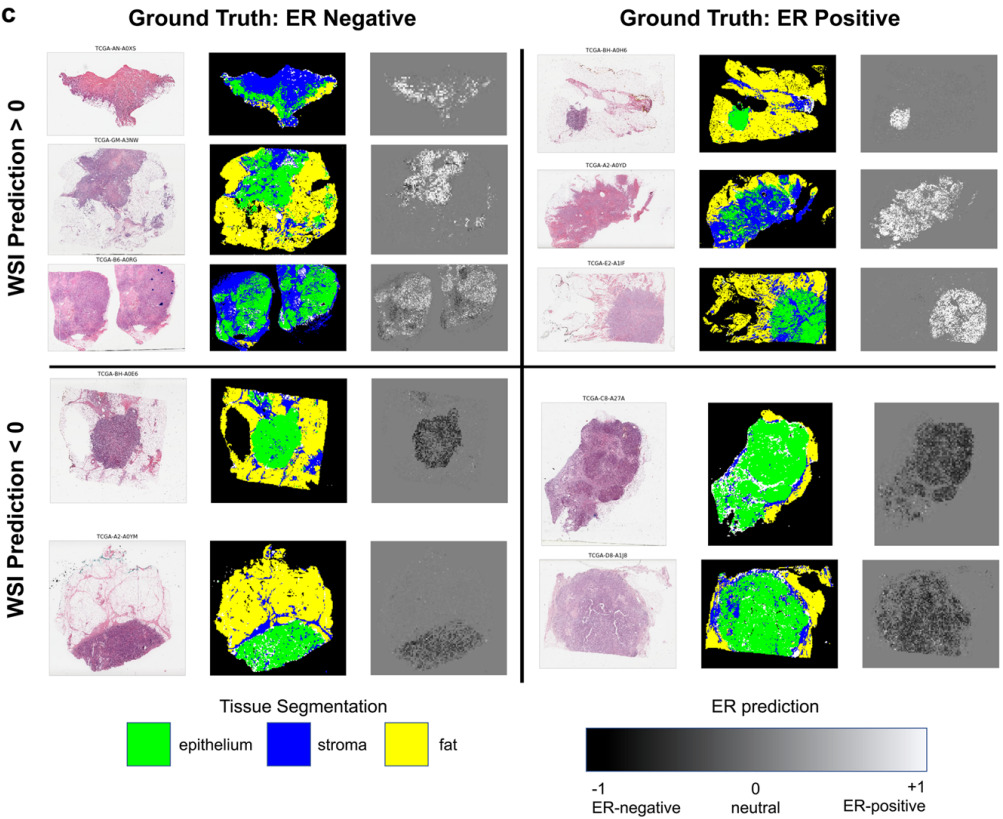
Protein biomarkers are a unique case because IHC slides of the same or adjacent tissue can be produced. Even if the model is trained with patient-level annotations, IHC can provide a means of qualitative validation. Shamai et al. explored this for breast cancer ER status [Shamai2019].
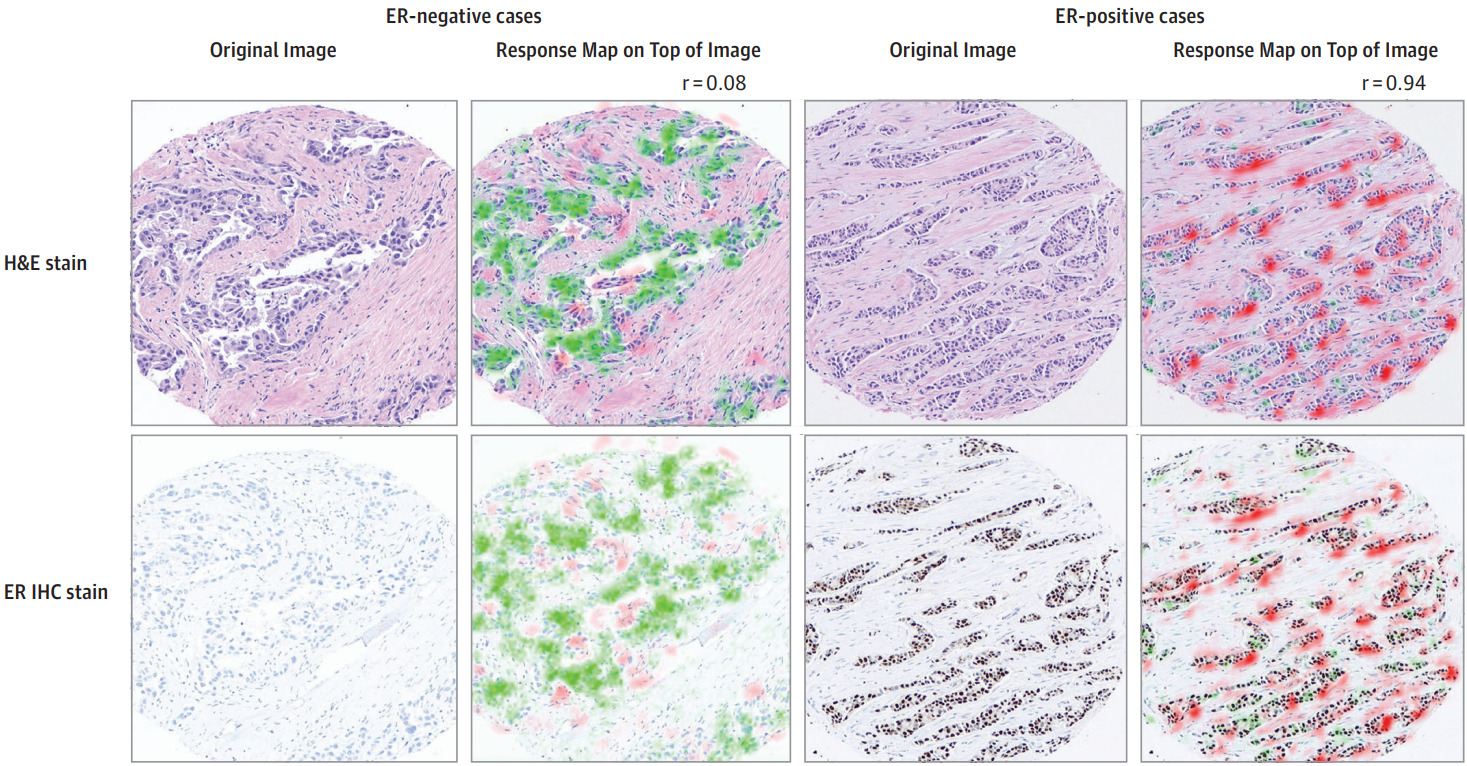
Spatial transcriptomics, where the spatial distribution of mRNA expression is measured across a tissue section, can provide another means for validation [He2020].
Magnification for Modeling
An open question from the above selection of weakly supervised CNN methods is the magnification level that should be used.
Cellular details are not visible in lower magnifications. Yet, with higher magnifications, a smaller region of tissue can be contained in a tile of a fixed pixel size, reducing the contextual information.
Many studies downsized image tiles to fit the input size of a particular CNN architecture. Others selected a lower magnification for computational efficiency.
Gamble et al. found success with 5x magnification [Gamble2021], while Wang et al. compared with higher magnifications and found 5x to generally underperform [Wang2021a]. Sirinukunwattana et al. also compared magnification levels and found 5x best in some cases [Sirinukunwattana2019].
There is not yet a clear consensus on what magnification level should be used for modeling. It may depend on the type of biomarker you’re predicting.
Learning with Pathologist-Driven Features
The above methods all focus on CNNs. Is there still a place for interpretable hand-crafted features and traditional classifiers in predicting molecular biomarkers?
Hand-Crafted Tissue Features
Sadhwani et al. compared a weakly supervised CNN with an interpretable histologic subtype approach for predicting tumor mutational burden of lung cancer [Sadhwani2021]. They also tried combining each with clinical variables, as well as a hybrid model that included all features.
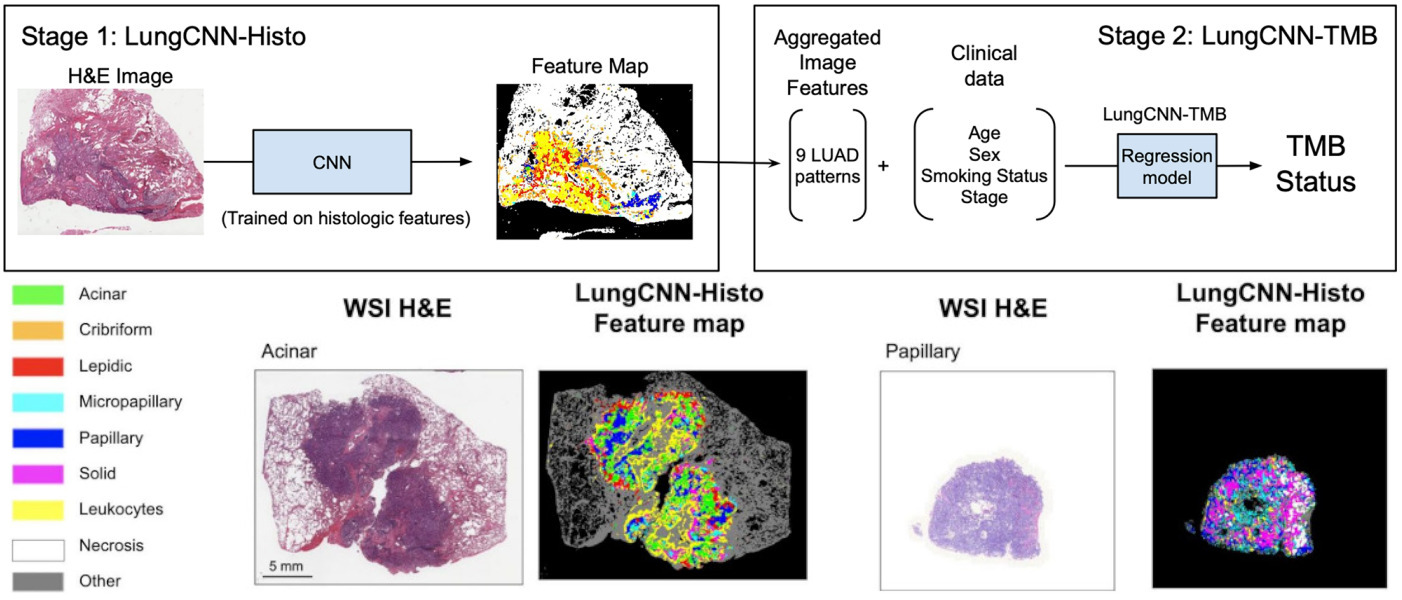
The hybrid model performed best, but some of the other variations were not far behind. These results demonstrate the potential of interpretable methods for tissue classification.
Diao et al. also focused on human-interpretable features but created them with deep learning [Diao2020]. They classified tissue and cell types with deep learning, then extracted features from each. These features were then aggregated across many images and mapped to biological concepts.
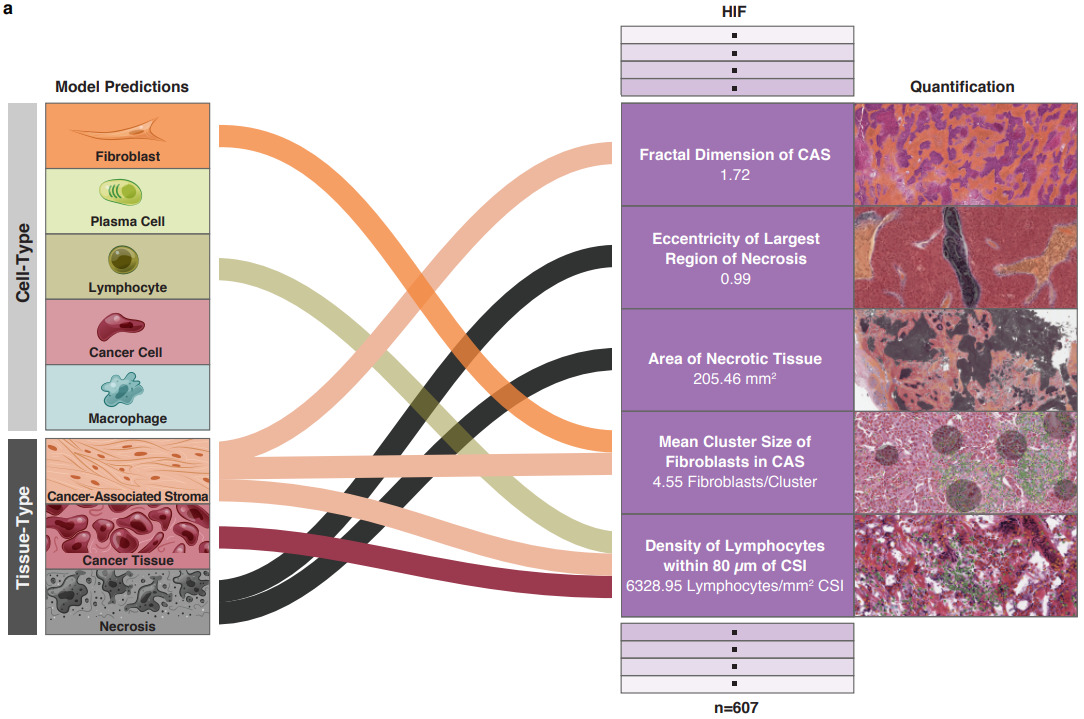
For example, the plots below show which feature and tissue types are associated with the PD-1 biomarker.
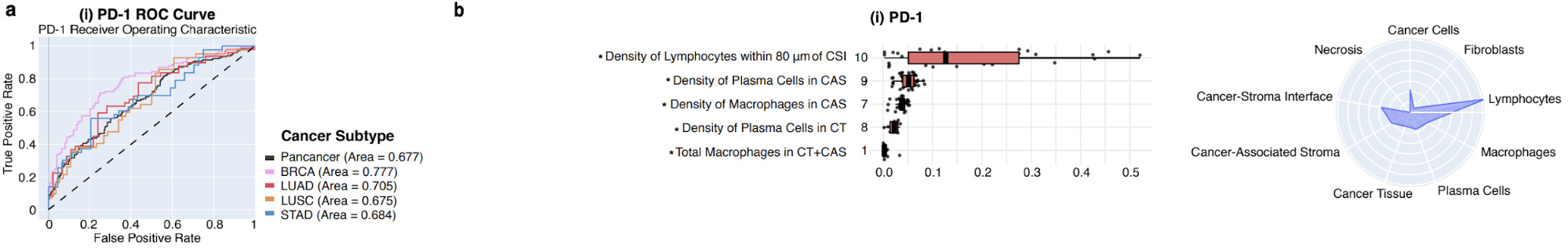
Hand-Engineered Features
As an alternative to hand-crafted features describing tissue morphology, Binder et al. evaluated hand-engineered features for breast cancer across a variety of molecular biomarkers: mutations, proteins, copy number variation, and DNA methylation [Binder2021].
They drew upon traditional computer vision features like intensity and gradient histograms, as well as SIFT, combined them in a bag of words framework, and used a kernel SVM for classification.
While the features selected don’t provide much insight from a morphological perspective, they paired them with spatial heatmaps to localize cancer cells and lymphocytes.
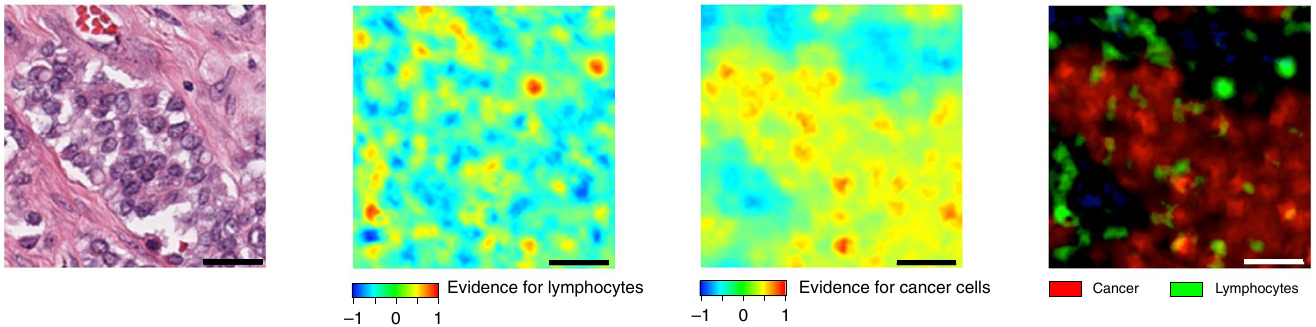
Mapping Cell Density
The above two methods open up the avenue for modeling the tissue microenvironment as an alternative – and more interpretable – means to predict molecular properties from WSIs.
Alghamdi et al. combined the classification of cells with modeling their spatial arrangement using deep learning [AlGhamdia2021]. They detected and classified all cells on the slide into five cell types. Then they formed a cell map for each type that identified the location of each cell. Compressing the cell maps by applying an averaging filter produced a smaller representation with each pixel representing the density of cells in that region. Finally, they trained a CNN on these 5D cell maps to predict receptor status.
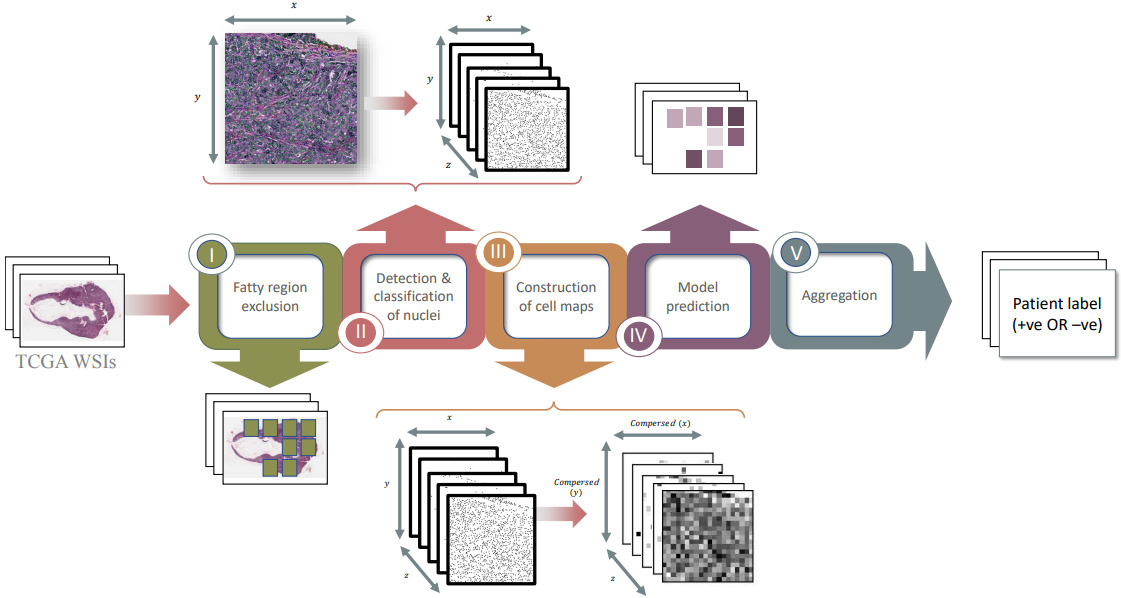
Experiments demonstrated that their model significantly outperforms one trained on raw H&E image tiles. The model also worked best when fatty regions are discarded.
Graphing Cellular Topology
An alternative method for mapping cells and representing spatial arrangements is with a graph neural network.
Lu et al. detected and classified nuclei across each WSI [Lu2020]. Each WSI can contain hundreds of thousands of nuclei, so they grouped neighboring nuclei into clusters. Then they formed a graph of cellular architecture by connecting neighboring clusters. They applied a graph convolutional network to predict HER2 and PR status of breast cancer, outperforming CNN methods operating in image tiles.
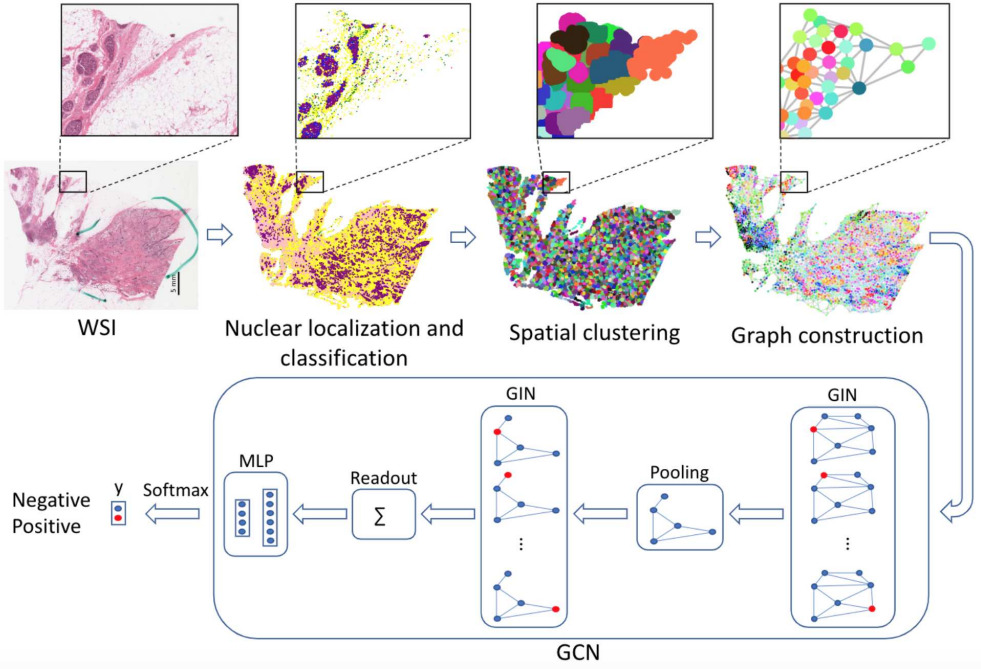
While fewer studies have modeled cell types, densities, distributions, and spatial arrangements for predicting molecular biomarkers, these approaches clearly present opportunities for future exploration.
Additional Considerations
While the bottom-up and pathologist-driven models discussed above cover the majority of machine learning research to predict molecular biomarkers, there are a few more approaches for specific scenarios and some additional considerations for modeling.
Strong Supervision
For the majority of molecular biomarkers, we only have patient- or slide-level labels. Protein markers assessed with immunohistochemistry (IHC) are an exception.
By staining serial sections or restaining a single section and aligning the WSIs, we can obtain localized ground truth annotations for breast cancer receptor status.
Gamble et al. obtained H&E slides co-registered with ER, PR, and HER2 IHC, and pathologists scored 512x512 pixel patches for each receptor [Gamble2021].
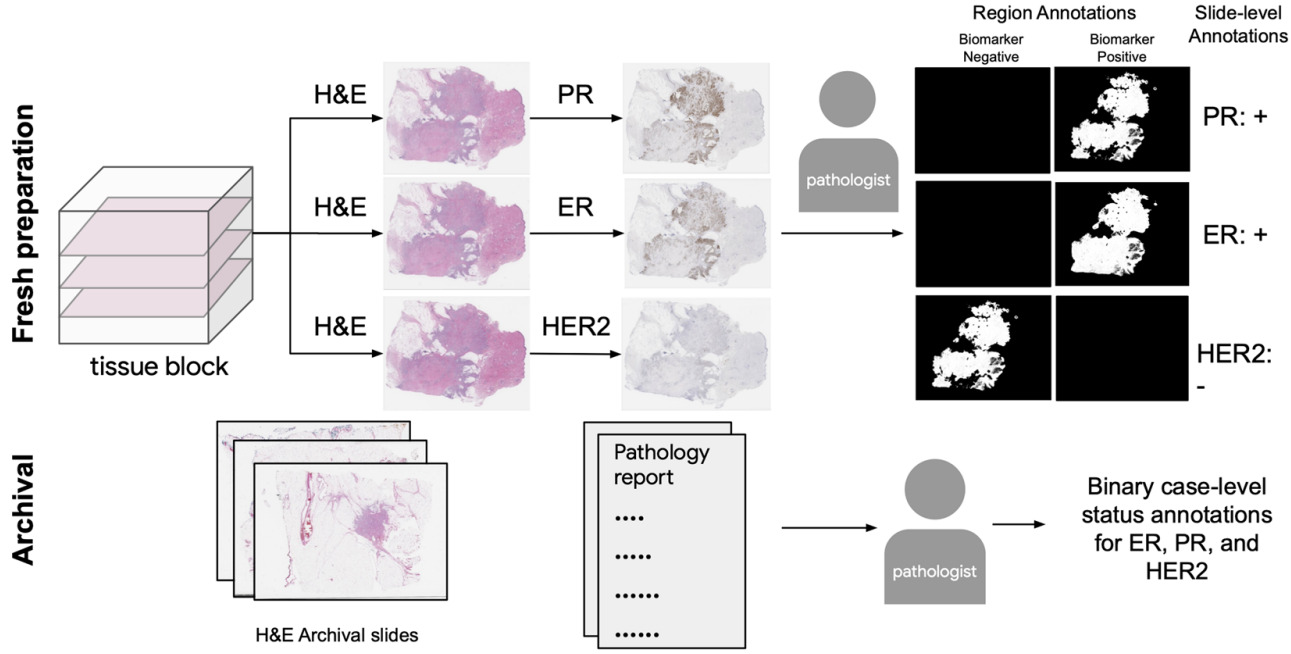
They trained a model for each receptor on these patches to predict positive, negative, or non-tumor. Slide-level predictions were made with a model that aggregates patch predictions.
Co-registered IHC/H&E images may be difficult to obtain but, when available, they provide very helpful annotations for predicting receptor status.
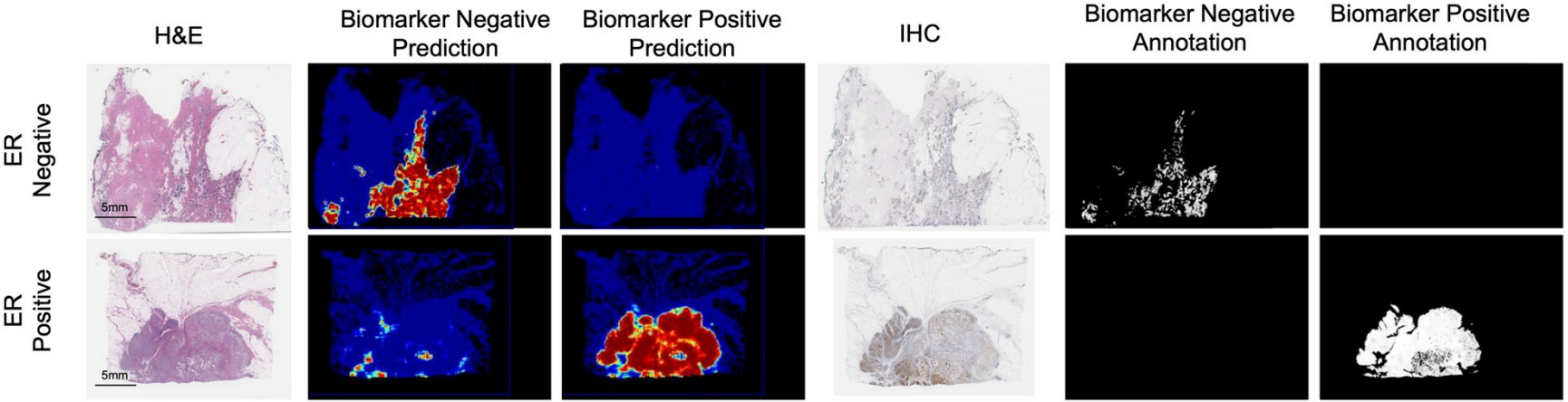
The annotations created by Gamble et al. are at the image patch level, however the status of individual cells can be observed with IHC.
Liu et al. created a cell-based deep learning method to predict Ki-67 status [Liu2020]. Pathologists annotated positive and negative Ki-67 regions on the IHC slides and labeled each cell within the regions as positive or negative. Then they transferred these labels to H&E and trained a CNN on 64x64 pixel patches.
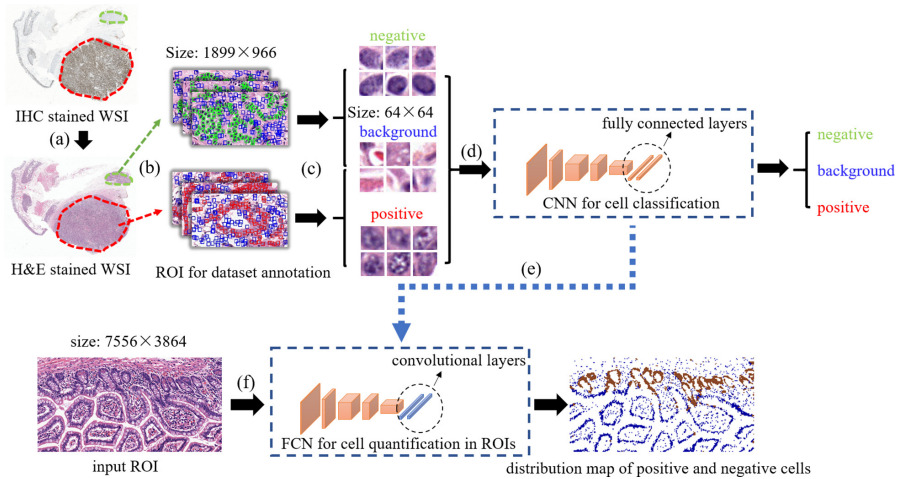
Their results demonstrated that tissue morphology has a close relation with molecular information.
Handling Small Datasets
When only a small number of patient samples is available for training, weakly supervised learning is much more challenging. The strongly supervised methods above are one possible solution. Another is through generating synthetic tiles.
Krause et al. trained a Conditional Generative Adversarial Network on their training set of colorectal cancer images to create new sample images with and without microsatellite instability [Krause2021]. Augmenting their training set with these synthetic images increased the size of their training set and improved model accuracy.
Pan-cancer Modeling
Most of the studies discussed above focus on a single or small set of cancer types and molecular properties.
Kather et al. tested their algorithm on WSIs from 5000 patients for 14 different tumor types [Kather2020]. They created a single deep learning workflow to predict point mutations, molecular and genomic subtypes, and hormone receptor status from H&E images.
Not every molecular property could be accurately predicted for every cancer type. But a large portion of them were shown to be significant.
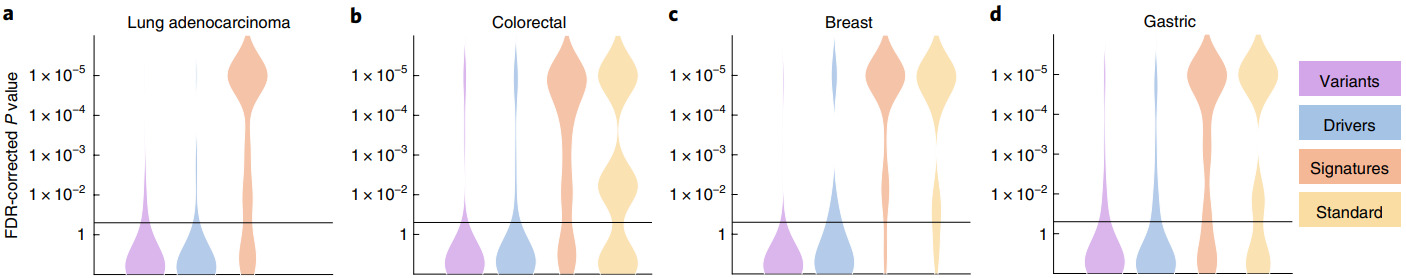
P-values for different cancer types and molecular properties including genetic variants, oncogenic drivers, high-level signatures, and standard-of-care features [Kather2020]
Fu et al. performed a similar test for predicting genomic, transcriptomic, and survival properties across 28 cancer types [Fu2020].
They used a relatively simple setup with transfer learning to extract a feature vector from each image tile. Then a linear classifier used the feature vectors to predict the class of each image tile, with the slide-level prediction computed as the average across tiles.
Even in this simpler setup without tuning the CNN for histology, they found links between tumor morphology and molecular composition in every cancer type and for almost every class of genomic and transcriptomic alteration.
Explainability
With these new prediction capabilities for molecular properties comes the need for techniques that can help pathologists determine the morphological differences between classes and identify potential model biases.
Seegerer et al. explored interpretable methods for predicting ER status from breast tumor images [Seegerer2020].
They used Layer-wise Relevance Propagation to locate relevant parts of the images for pathologists to examine. Stroma was found important for indicating ER+, while lymphocyte infiltration and high grade were associated with ER-. A larger study will be needed to further validate these observations.
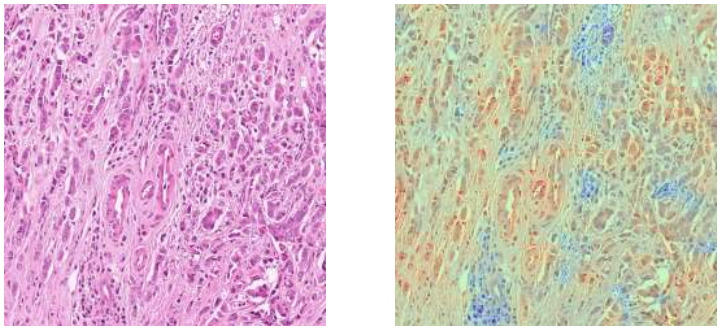
Layer-wise Relevance Propagation is just one of many pixel attribution methods. GradCam is also frequently used to produce this type of heatmap [Bychkov2021].
Alternatively, classifier predictions on smaller patches can be used to produce a class heatmap.
The heatmap for microsatellite instability shown above is one successful example [Saillard2021]. Gamble et al.’s results for receptor status is another [Gamble2021].
Rawat et al’s fingerprinting technique also presented helpful heatmaps to receptor status predictions [Rawat2020].
Biomarker Heterogeneity and Outcomes
Localized predictions provide an additional benefit in understanding tissue heterogeneity.
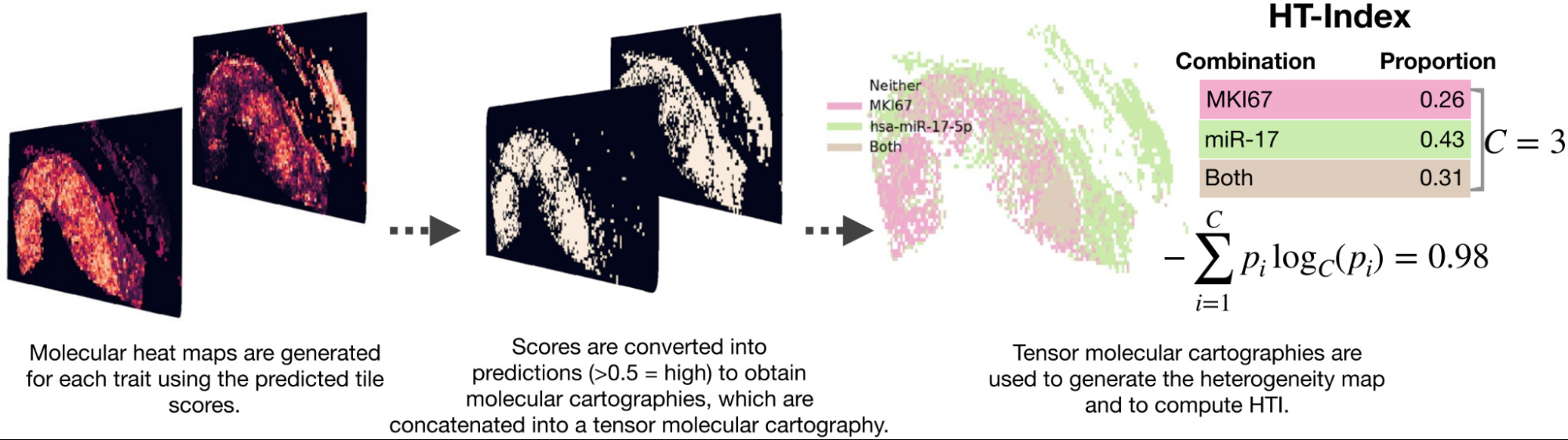
Levy-Jurgenson et al. developed a model to predict mRNA and miRNA expression from WSIs [Levy-Jurgenson2020]. The heatmaps produced are interesting, which they refer to as molecular cartography. But even more compelling is how they were able to quantify heterogeneity.
Examining a small set of genes at a time, they displayed the model predictions over the slide, coloring each region based on the predictions. In the case of 2 genes, there were distinct colors for positive/positive, positive/negative, and negative/positive. To quantify heterogeneity, they computed the fraction of the slide belonging to each group and applied Shannon’s entropy formula. High heterogeneity was found to be linked with poor survival, especially for breast cancer.
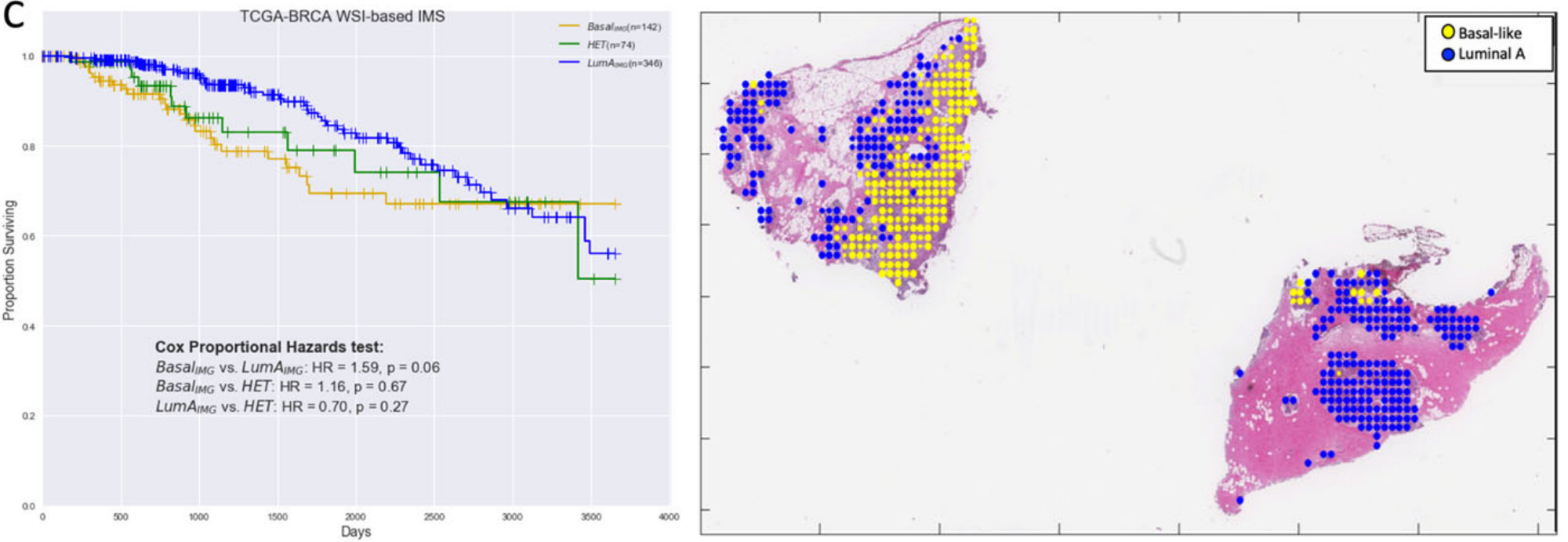
Jaber et al. used deep learning to predict Basal versus Luminal A genomic subtypes of breast cancer [Jaber2020]. The image-based biomarkers were found to be more predictive of patient survival than the molecular subtypes themselves. Further, the heterogeneous samples – predicted to contain both Basal and Luminal A subtypes – had an intermediate prognosis.
Bychkov et al. also demonstrated that their image-based model can predict patient outcomes [Bychkov2021]. They trained their model to predict breast cancer HER2 status and demonstrated that these predictions were also associated with survival and treatment response.
Validation and Generalizability
Finally, let’s look at two of the most important aspects of a machine learning model: validation and generalizability.
There are methods to improve biomarker classifier performance, but what variables affect model accuracy?
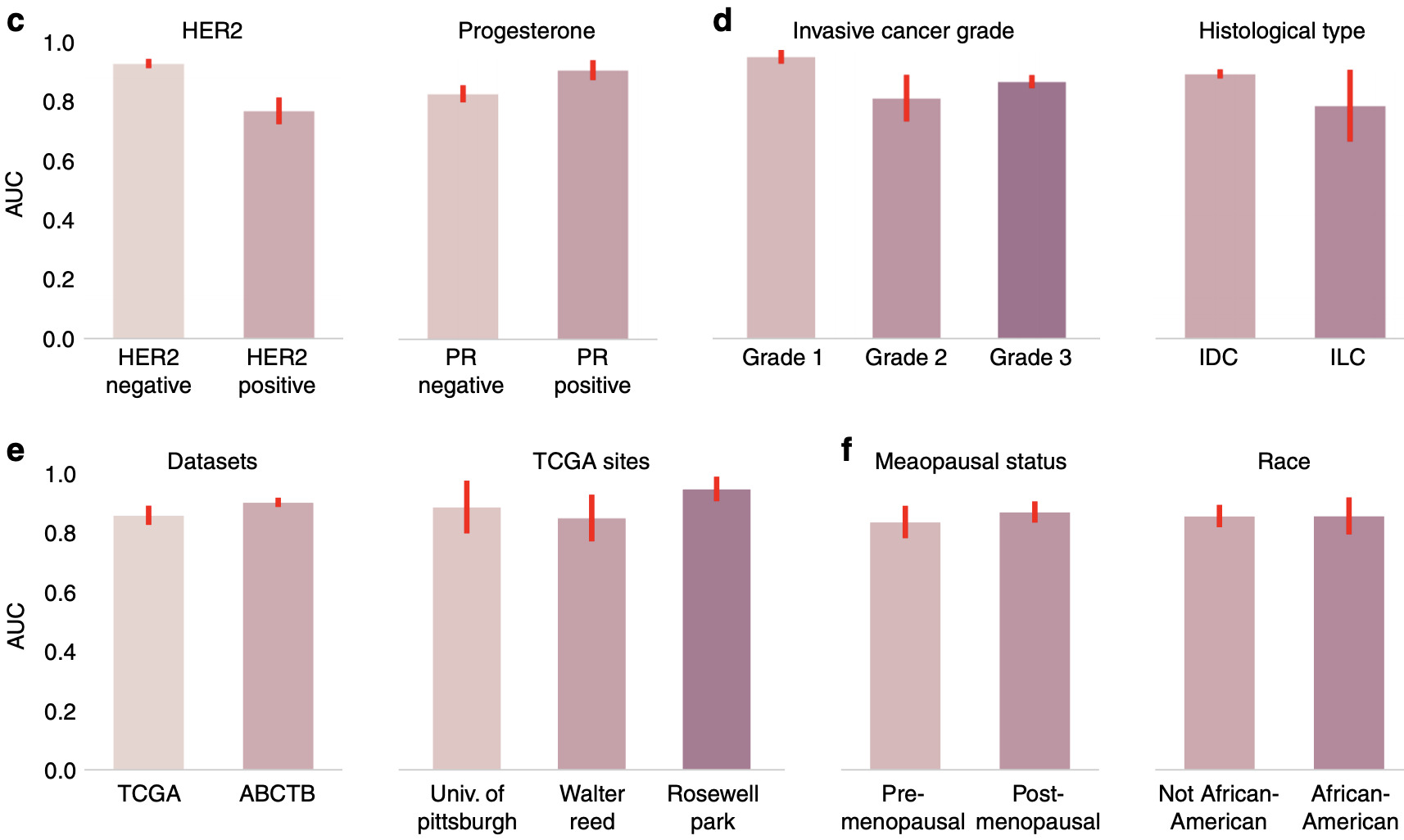
Naik et al. calculated model performance for a number of different divisions of histological and clinical variables [Naik2020]. Some affected model performance significantly more than others.
This method of analyzing model performance is critical to understanding how well it generalizes and where improvements could be made in the future. It can also identify hidden batch effects [Schmitt2020].
Some of the studies discussed in this article assess performance on an independent cohort, while others simply split a single dataset into training, validation, and test sets.
Those that do test on an independent cohort typically see a performance drop because the test set was collected at a different hospital, processed in a different lab, or imaged with a different scanner.
Hohne et al. found their model performed well for predicting BRAF mutation status for thyroid cancer but performance dropped about 10% when tested on two held out sets and even further for NTRK mutation status [Hohne2021].
Sirinukunwattana et al. took steps to improve generalization performance by applying adversarial domain adaptation to encourage their model to learn domain-invariant features [Sirinukunwattana2019].
Alternatively, Rawat et al. accommodated variations in staining by different labs using a GAN-based style transfer [Rawat2020]. A large portion of the improvement in their model was attributed to this step.
For additional ideas to improve model generalizability, see my article on improving robustness to domain shifts.
Recommendations
So what is the best approach for predicting molecular biomarkers from H&E WSIs?
I don’t think there is a single best solution.
The cell map and graph neural network approaches based on detected and classified nuclei can provide a powerful and somewhat interpretable means to characterize the tumor microenvironment.
But they are also based on a pathologist’s view of important tissue characteristics. Again, great for interpretability, but perhaps with limited power to distinguish tissue classes that are too complex for a pathologist to identify visually.
The best alternative is CNN-based models using a weakly supervised attention model. Though if strong supervision is possible – for example, from IHC – such a model is preferred.
If an end-to-end model is used, a random subset of tiles must be selected during each epoch in order to fit on the GPU.
Self-supervision is most beneficial when your data is significantly different from standard benchmark datasets like ImageNet, and you have a large amount of unlabeled or weakly labeled data from which to learn a representation.
By pretraining with self-supervised learning, the model can compress image tiles before applying an attention model to the whole slide. This approach has shown great results and even seems to generalize to new datasets better than alternative models.
Insights into intratumoral heterogeneity and associations with patient outcomes also demonstrate the powerful capability of these methods.
The black box nature of deep learning presents challenges for medical applications. More explainable methods would build trust, identify potential biases, and perhaps teach pathologists what characteristics of tumor morphology these powerful models are using to predict molecular biomarkers.
Finally, more rigorous validation is needed on larger data sets with a wider variety of patients. When applied to multiple cohorts of patients, the domain shift due to processing in different labs with different populations of patients also becomes a challenge. Tissue staining methods can vary from lab to lab and can change over time, and tumors from different populations of patients may have different characteristics. A variety of techniques are available to mitigate performance drops due to domain shifts, for validation is still essential.
Deep learning-based biomarkers from H&E could provide an additional tool for pathologists and new insights for companion diagnostics and drug discovery. Based on only H&E images and no longer limited by the amount of tissue excised or the processing time for individual molecular tests, deep learning-based methods can provide easier access to vital information about a tumor. They may yield a screening method to determine when to run molecular tests or an alternative when molecular tests are not possible.
Does your team need a better strategy for deep learning-based histology biomarkers?
I’ve worked with a variety of teams to develop advanced machine learning algorithms to extract new insights from pathology images. Schedule a free Machine Learning Strategy Session to get started.
References
[Abbasi-Sureshjani2021] S. Abbasi-Sureshjani, A. Yüce, S. Schönenberger, M. Skujevskis, U. Schalles, F. Gaire, K. Korski, Molecular Subtype Prediction for Breast Cancer Using H&E Specialized Backbone (2021), MICCAI Workshop on Computational Pathology
[AlGhamdia2021] H.M. AlGhamdiă, N.A. Koohbanani, N. Rajpoot, S.E.A. Raza, A Novel Cell Map Representation for Weakly Supervised Prediction of ER and PR Status from H&E WSIs (2021), MICCAI Workshop on Computational Pathology
[Anand2021] D. Anand, K. Yashashwi, N. Kumar, S. Rane, P.H. Gann, A. Sethi, Weakly supervised learning on unannotated hematoxylin and eosin stained slides predicts BRAF mutation in thyroid cancer with high accuracy (2021), Journal of Pathology
[Binder2021] A. Binder, M. Bockmayr, M. Hägele, S. Wienert, D. Heim, K. Hellweg, F. Klauschen, Morphological and molecular breast cancer profiling through explainable machine learning (2021), Nature Machine Intelligence
[Bychkov2021] D. Bychkov, N. Linder, A. Tiulpin, H. Kücükel, M. Lundin, S. Nordling, H. Sihto, J. Isola, T. Lehtimäki, P.L. Kellokumpu-Lehtinen. K. von Smitten, Deep learning identifies morphological features in breast cancer predictive of cancer ERBB2 status and trastuzumab treatment efficacy (2021), Scientific Reports
[Coudray2018] N. Coudray, P.S. Ocampo, T. Sakellaropoulos, N. Narula, M. Snuderl, D. Fenyö, A.L. Moreira, N. Razavian, A. Tsirigos, Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning (2018), Nature Medicine
[Couture2018a] H.D. Couture, L.A. Williams, J. Geradts, S.J. Nyante, E.N. Butler, J.S. Marron, C.M. Perou, M.A. Troester, M. Niethammer, Image analysis with deep learning to predict breast cancer grade, ER status, histologic subtype, and intrinsic subtype (2018), NPJ Breast Cancer
[Couture2018b] H.D. Couture, J.S. Marron, C.M. Perou, M.A. Troester, M. Niethammer M, Multiple Instance Learning for Heterogeneous Images: Training a CNN for Histopathology (2018), Medical Image Computing and Computer-Assisted Intervention
[Diao2020] J.A. Diao, W.F. Chui, J.K. Wang, R.N. Mitchell, S.K. Rao, M.B. Resnick, A. Lahiri, C. Maheshwari, B. Glass, V. Mountain, J.K. Kerner, Dense, high-resolution mapping of cells and tissues from pathology images for the interpretable prediction of molecular phenotypes in cancer (2020), bioRxiv
[Fu2020] Y. Fu, A.W. Jung, R.V. Torne, S. Gonzalez, H. Vöhringer, A. Shmatko, L.R. Yates, M. Jimenez-Linan, L. Moore, M. Gerstung, Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis (2020), Nature Cancer
[Gamble2021] P. Gamble, R. Jaroensri, H. Wang, F. Tan, M. Moran, T. Brown, I. Flament-Auvigne, E.A. Rakha, M. Toss, D.J. Dabbs, P. Regitnig, Determining breast cancer biomarker status and associated morphological features using deep learning (2021), Communications Medicine
[He2020] B. He, L. Bergenstråhle, L. Stenbeck, A. Abid, A. Andersson, A. Borg, J. Maaskola, J. Lundeberg, J. Zou, Integrating spatial gene expression and breast tumour morphology via deep learning (2020), Nature Biomedical Engineering
[Hohne2021] J. Höhne, J. de Zoete, A.A. Schmitz, T. Bal, E. di Tomaso, M. Lenga, Detecting genetic alterations in BRAF and NTRK as oncogenic drivers in digital pathology images: towards model generalization within and across multiple thyroid cohorts (2021), MICCAI Workshop on Computational Pathology
[Ilse2018] M. Ilse, J. Tomczak, M. Welling, Attention-based Deep Multiple Instance Learning (2018), Proceedings of the International Conference on Machine Learning
[Jaber2020] M.I. Jaber, B. Song, C. Taylor, C.J. Vaske, S.C. Benz, S. Rabizadeh, P. Soon-Shiong, C.W. Szeto, A deep learning image-based intrinsic molecular subtype classifier of breast tumors reveals tumor heterogeneity that may affect survival (2020), Breast Cancer Research
[Kather2019a] J.N. Kather, A.T. Pearson, N. Halama, D. Jäger, J. Krause, S.H. Loosen, A. Marx, P. Boor, F. Tacke, U.P. Neumann, H.I. Grabsch, Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer (2019), Nature Medicine
[Kather2019b] J.N. Kather, J. Schulte, H.I. Grabsch, C. Loeffler, H. Muti, J. Dolezal, A. Srisuwananukorn, N. Agrawal, S. Kochanny, S. von Stillfried, P. Boor, Deep learning detects virus presence in cancer histology (2019), bioRxiv
[Kather2020] J.N. Kather, L.R. Heij, H.I. Grabsch, C. Loeffler, A. Echle, H.S. Muti, J. Krause, J.M. Niehues, K.A. Sommer, P. Bankhead, L.F. Kooreman, Pan-cancer image-based detection of clinically actionable genetic alterations (2020), Nature Cancer
[Kim2020] R.H. Kim, S. Nomikou, N. Coudray, G. Jour, Z. Dawood, R. Hong, E. Esteva, T. Sakellaropoulos, D. Donnelly, U. Moran, A. Hatzimemos, A Deep Learning Approach for Rapid Mutational Screening in Melanoma (2020), bioRxiv
[Krause2021] J. Krause, H.I. Grabsch, M. Kloor, M. Jendrusch, A. Echle, R.D. Buelow, P. Boor, T. Luedde, T.J. Brinker, C. Trautwein, A.T. Pearson, Deep learning detects genetic alterations in cancer histology generated by adversarial networks (2021), Journal of Pathology
[LaBarbera2020] D. La Barbera, A. Polónia, K. Roitero, E. Conde-Sousa, V. Della Mea, Detection of HER2 from Haematoxylin-Eosin Slides Through a Cascade of Deep Learning Classifiers via Multi-Instance Learning (2020), Journal of Imaging
[Levy-Jurgenson2020] A. Levy-Jurgenson, X. Tekpli, V.N. Kristensen, Z. Yakhini, Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer (2020), Scientific Reports
[Liu2020] Y. Liu, X. Li, A. Zheng, X. Zhu, S. Liu, M. Hu, Q. Luo, H. Liao, M. Liu, Y. He, Y. Chen, Predict Ki-67 Positive Cells in H&E-Stained Images Using Deep Learning Independently From IHC-Stained Images (2020), Frontiers in Molecular Biosciences
[Lu2020] W. Lu, S. Graham, M. Bilal, N. Rajpoot, F. Minhas, Capturing Cellular Topology in Multi-Gigapixel Pathology Images (2020), Conference on Computer Vision and Pattern Recognition Workshops
[Naik2020] N. Naik, A. Madani, A. Esteva, N.S. Keskar, M.F. Press, D. Ruderman, D.B. Agus, R. Socher, Deep learning-enabled breast cancer hormonal receptor status determination from base-level H&E stains (2020), Nature Communications
[Popovici2017] V. Popovici, E. Budinská, L. Dušek, M. Kozubek, F. Bosman, Image-based surrogate biomarkers for molecular subtypes of colorectal cancer (2017), Bioinformatics
[Rawat2020] R.R. Rawat, I. Ortega, P. Roy, F. Sha, D. Shibata, D. Ruderman, D.B. Agus, Deep learned tissue “fingerprints” classify breast cancers by ER/PR/Her2 status from H&E images (2020), Scientific Reports
[Rymarczyk2020] D. Rymarczyk, A. Borowa, J. Tabor, B. Zieliński, Kernel Self-Attention in Deep Multiple Instance Learning (2020), arXiv
[Sadhwani2021] A. Sadhwani, H.W. Chang, A. Behrooz, T. Brown, I. Auvigne-Flament, H. Patel, R. Findlater, V. Velez, F. Tan, K. Tekiela, E. Wulczyn, Comparative analysis of machine learning approaches to classify tumor mutation burden in lung adenocarcinoma using histopathology images (2021), Scientific Reports
[Saillard2021] C. Saillard, O. Dehaene, T. Marchand, O. Moindrot, A. Kamoun, B. Schmauch, S. Jegou, Self supervised learning improves dMMR/MSI detection from histology slides across multiple cancers (2021), arXiv
[Schirris2021] Y. Schirris, E. Gavves, I. Nederlof, H.M. Horlings, J. Teuwen, DeepSMILE: Self-supervised heterogeneity-aware multiple instance learning for DNA damage response defect classification directly from H&E whole-slide images (2021), arXiv
[Schmauch2020] B. Schmauch, A. Romagnoni, E. Pronier, C. Saillard, P. Maillé, J. Calderaro, A. Kamoun, M. Sefta, S. Toldo, M. Zaslavskiy, T. Clozel, A deep learning model to predict RNA-Seq expression of tumours from whole slide images (2020), Nature Communications
[Schmitt2020] M. Schmitt, R.C. Maron, A. Hekler, A. Stenzinger, A. Hauschild, M. Weichenthal, M. Tiemann, D. Krahl, H. Kutzner, J.S. Utikal, S. Haferkamp, Hidden Variables in Deep Learning Digital Pathology and Their Potential to Cause Batch Effects: Prediction Model Study (2021), Journal of Medical Internet Research
[Seegerer2020] P. Seegerer, A. Binder, R. Saitenmacher, M. Bockmayr, M. Alber, P. Jurmeister, F. Klauschen, K.R. Müller, Interpretable Deep Neural Network to Predict Estrogen Receptor Status from Haematoxylin-Eosin Images (2020), Artificial Intelligence and Machine Learning for Digital Pathology
[Shamai2019] G. Shamai, Y. Binenbaum, R. Slossberg, I. Duek, Z. Gil, R. Kimmel,Artificial Intelligence Algorithms to Assess Hormonal Status From Tissue Microarrays in Patients With Breast Cancer (2019), JAMA Network Open
[Sirinukunwattana2019] K. Sirinukunwattana, E. Domingo, S. Richman, V.H. Koelzer, Image-based consensus molecular subtype classification (imCMS) of colorectal cancer using deep learning (2019), bioRxiv
[Wang2021a] X. Wang, C. Zou, Y. Zhang, X. Li, C. Wang, F. Ke, J. Chen, W. Wang, D. Wang, X. Xu, L. Xie, Prediction of BRCA Gene Mutation in Breast Cancer Based on Deep Learning and Histopathology Images (2021), Frontiers in Genetics
[Wang2021b] Y. Wang, K. Kartasalo, P. Weitz, B. Acs, M. Valkonen, C. Larsson, P. Ruusuvuori, J. Hartman, M. Rantalainen, Predicting molecular phenotypes from histopathology images: a transcriptome-wide expression-morphology analysis in breast cancer (2021), Cancer Research
[Xu2020] H. Xu, S. Park, S.H. Lee, T.H. Hwang, Using transfer learning on whole slide images to predict tumor mutational burden in bladder cancer patients (2019), bioRxiv